
Classification of mitochondrial diseases
13 kinds of protein required for the main functions of a mitochondria are translated from mtDNA. mtDNA abnormalities decrease mitochondrial function and prevent energy supply, eventually causing mitochondrial diseases.
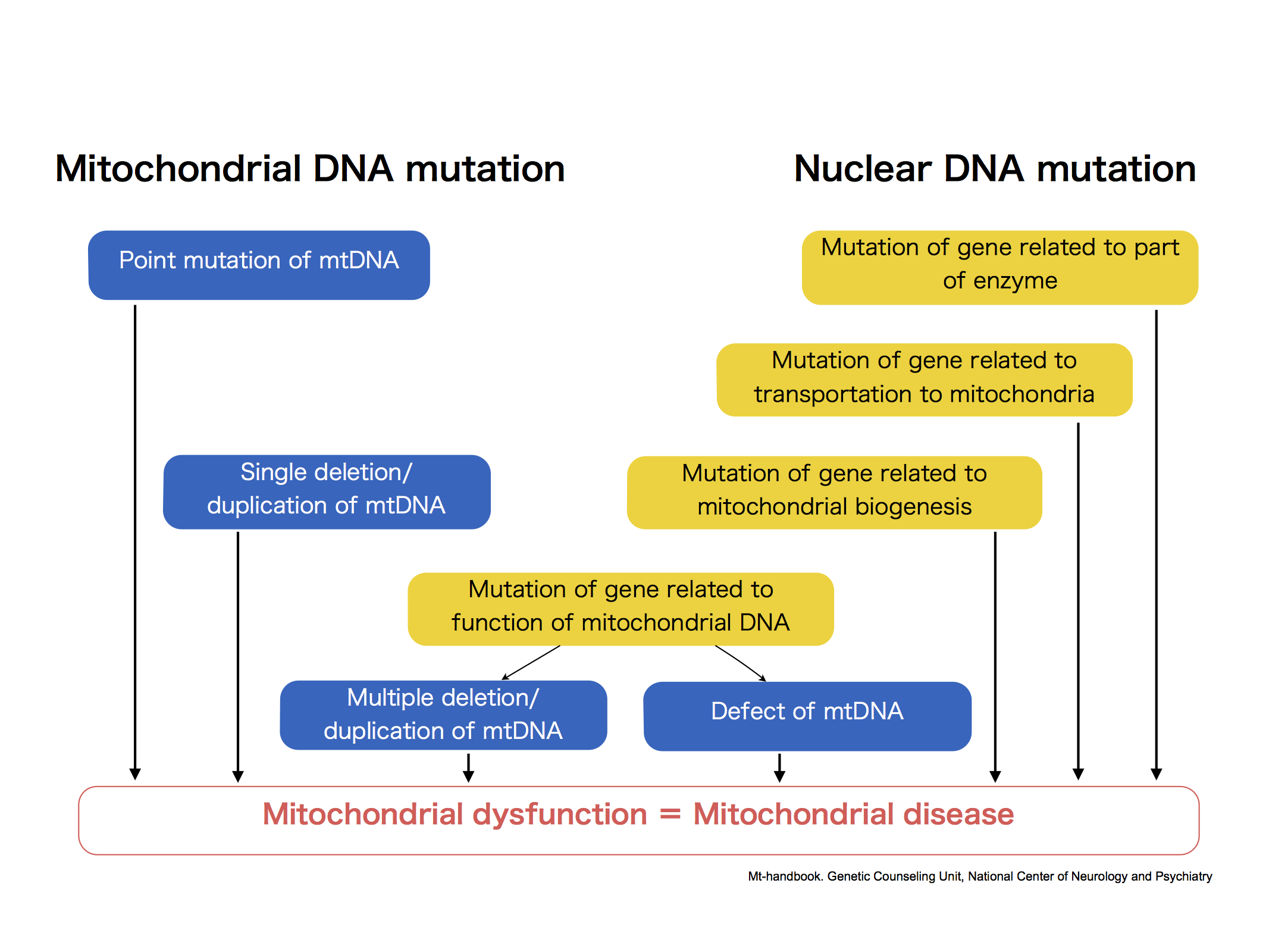
Also, approximately 1500 kinds of proteins required for mitochondria are constructed based on the information in nuclear DNA. Moreover, the nuclear DNA involves in the function of mtDNA itself. Therefore, abnormalities in nuclear DNA cause wide and various harmful effect on energy metabolic pathway (from decomposition and synthesis of substances to energy production in respiratory chain complexes), and result in many types of mitochondrial disease.
Diseases that are mainly caused by mitochondrial DNA abnormalities
| Name | Symptoms |
|---|---|
| CPEO: Chronic Progressive External Ophthalmoplegia http://omim.org/entry/157640 |
<mtDNA Abnormalities>In CPEO, external eye muscles are paralyzed and patients are unable to move the eyes. When complicated by retinopathy and cardiac conduction disturbances, CPEO is called Kearns-Sayre Syndrome. |
| Pearson Syndrome http://omim.org/entry/557000 |
<mtDNA Abnormalities>Pearson syndrome patients develop sideroblastic anemia and exocrine pancreatic insufficiency in infancy. Patients who survive childhood develop Kearns-Sayre Syndrome. |
| KSS: Kearns-Sayre Syndrome http://omim.org/entry/530000 |
<mtDNA Abnormalities>KSS is a syndrome characterized by ptosis and external ophthalmoplegia, cardiac conduction disturbance, and retinitis pigmentosa. Symptoms usually develop in childhood. Because the etiology of the disease is extremely similar to that of CPEO, KSS is considered a severe form of CPEO. Abnormal mtDNA is observed in 70% of CPEO and 90% of KSS patients. |
| DAD: Diabetes and Deafness http://omim.org/entry/520000 |
<mtDNA Abnormalities>In DAD, patients develop diabetes with hearing impairment. |
| LHON: Leber Hereditary Optic Neuropathy http://omim.org/entry/535000 |
<mtDNA Abnormalities>LHON patients develop bilateral blurred vision, mainly from ages 10–30 years old. Subsequently, optic disk pallor begins gradually and usually results in optic atrophy in around one year. |
| LHON-Plus http://omim.org/entry/500001 |
<mtDNA Abnormalities>In addition to optic neuropathies of LHON, patients develop CNS symptoms such as unusual motion, impaired coordination, dystonia, ataxia, spasticity, and cardiac disorders. |
| NARP: Neuropathy, Ataxia, and Retinitis Pigmentosa Syndrome http://omim.org/entry/551500 |
<mtDNA Abnormalities>In NARP, patients develop neuropathy, ataxia, and retinitis pigmentosa. This syndrome has a strong association with MILS. |
| MILS: Maternally-Inherited Leigh Syndrome http://omim.org/entry/256000 |
<mtDNA Abnormalities>MILS is a type of Leigh's disease that results from mtDNA abnormalities. |
| MELAS: Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes http://omim.org/entry/540000 |
<mtDNA Abnormalities>MELAS is a mitochondrial disease accompanied by stroke-like symptoms, such as rapid deterioration of consciousness and palsy. Symptoms may occur in organs such as the heart, spleen, and ears or endocrine organs. The type and degree of symptoms vary between patients, and a patient’s symptoms may change over time. |
| MERRF: Myoclonic Epilepsy and Ragged-Red Fiber Disease http://omim.org/entry/545000 |
<mtDNA Abnormalities>MERRF is a mitochondrial disease accompanied by myoclonus, and symptoms occur mainly in the brain and muscles. Symptoms include shaky body (cerebellar symptoms) and involuntary muscle movements (myoclonus). |
| FBSN: Familial Bilateral Striatal Necrosis/Striatonigral Degeneration http://omim.org/entry/500003 |
<mtDNA Abnormalities>FBSN results from a mitochondrial respiratory chain complex V (ATP synthetase) deficiency that can be attributed to mtDNA abnormalities. |
| Luft Disease http://omim.org/entry/251900 |
<mtDNA Abnormalities>Luft disease is a mitochondrial myopathy that is often accompanied by fever, profuse sweating, polyphagia, polydipsia, ragged red fibers, tachycardia, and hypermetabolism. Patients develop mild weakness and exercise intolerance. |
| AID: Aminoglycoside-induced Deafness http://omim.org/entry/580000 |
<mtDNA Abnormalities>In AID, complete deafness is caused by aminoglycoside drugs. |
| Multiple Deletions of Mitochondrial DNA http://www.ncbi.nlm.nih.gov/books/NBK1203/ |
<mtDNA Abnormalities>Multiple deletions of mitochondrial DNA syndromes are caused by multiple genetic defects of mtDNA (at least 5 nuclear genes are known). Clinical symptoms are usually similar to CPEO. Related symptoms include hearing loss, myopathy, ataxia, peripheral neuropathy, dysarthria, optic atrophy, dementia, and parkinsonism, among others. |
Diseases that are mainly caused by nuclear DNA Abnormalities
| Name | Symptoms |
|---|---|
| Alpers syndrome, Progressive infantile poliodystrophy http://omim.org/entry/203700 |
<Defects in mitochondrial DNA maintenance> These patients develop seizures, dementia, spasticity, blindness, hepatic dysfunction, and brain degeneration. This syndrome is caused by a nuclear DNA mutation (POLG gene: mtDNAreplicase), and this is the most severe phenotype of POLG gene mutation. |
| MIRAS: Mitochondrial Recessive Ataxia Syndrome http://omim.org/entry/607459 |
<Defects in mtDNA maintenance> Together with SANDO, MIRAS is now classified as a POLG-related disease on the Ataxia Neuropathy Spectrum (ANS). |
| MNGIE: Mitochondrial Neurogastrointestinal Encephalomyopathy http://omim.org/entry/603041 |
<Defects in mtDNA maintenance> In MINGE, defects in thymidine phosphorylase cause abnormalities in nucleic acid metabolism important for DNA synthesis. Though the numbers of dividing cells are small, these abnormalities greatly affect the quantity and quality of mtDNA because mtDNA replication occurs frequently. Patients develop CPEO, a type of MTDPS, peripheral neuropathy, GI disorders, leukoencephalopathy, cachexia (extreme exhaustion), lactic acidosis, and red rag fibers, among a host of other diseases. This disease arises due to the nuclear DNA mutations in the TYMP gene, which encodes thymidine phosphorylase. |
| MTDPS: Mitochondrial DNA Depletion Syndrome http://omim.org/entry/603041 http://omim.org/entry/251880 |
<Defects in mtDNA maintenance> MTDPS develops due to abnormalities of genes involved in the production of components for mtDNA and has various symptoms. This disease is a major cause of myopathy in infancy and childhood, as well as of liver and brain disorders. |
| POLG-Related Disorders (POLG mutation) http://omim.org/entry/174763 http://omim.org/entry/613662 http://omim.org/entry/604983 http://omim.org/entry/203700 http://omim.org/entry/157640 http://omim.org/entry/258450 http://omim.org/entry/607459 http://omim.org/entry/606075 http://omim.org/entry/609283 http://omim.org/entry/609286 http://omim.org/entry/610131 http://omim.org/entry/603041 http://omim.org/entry/604419 |
<Defects in mtDNA maintenance>
|
| SANDO: Sensory Ataxia Neuropathy Dysarthria Ophthalmoplegia http://omim.org/entry/174763 |
<Abnormalities in mtDNA maintenance> Together with MIRAS, this syndrome is now classified as a POLG-related disease on the Ataxia neuropathy spectrum (ANS) |
| LBSL: Leukoencephalopathy with Brainstem and Spinal cord involvement and Lactate elevation http://omim.org/entry/611105 |
<Disorders in translation of mtDN> LBSL is a brainstem and spinal cord lesion caused by mitochondrial aspartyl-tRNA synthetase deficiency. Patients develop high lactic acid levels, muscular atrophy, muscle weakness, ataxia, and tremors. This disease is caused by abnormalities in nuclear DNA, particularly in the DARS2 gene. |
| Co-Enzyme Q10 Deficiency http://omim.org/entry/607426 |
<Lipid membrane abnormalities> These patients develop encephalomyopathy, mental retardation, exercise intolerance, red rag fibers, and recurrent myoglobinuria. This disease can be attributed to nuclear DNA abnormalities. |
| Leigh Disease or Syndrome http://omim.org/entry/256000 http://omim.org/entry/308930 |
<Mainly due to respirator chain compex abnormalities> This disease is characterized by symptoms, such as psychomotor retardation, that begin at 2 years old and present with high levels of lactic acid and pyruvic acid in the blood, as well as symmetrical necrotic lesions in the brainstem and basal ganglia confirmed by CT and MRI. The most common initial presentation is feeding disturbances, followed by delayed motor development and hypotonia of muscles and body cavities. Patients develop eye symptoms, such as oculomotor abnormalities, optic atrophy, and visual impairment, and they also develop seizures, respiratory compromise, and cerebellar symptoms. At least 26 gene abnormalities have been identified, and those abnormalities are related to pyruvate dehydrogenase (PDHC) deficiency and respiratory chain enzymes (complex I, II, IV, and V) deficiencies. This syndrome is caused by nuclear DNA abnormalities. |
| Complex I Deficiency http://omim.org/entry/252010 |
<Respiratory chain complex abnormalities> Low activity of mitochondrial respiratory chain complex I (NADH -ubiquinone oxidoreductase) decreases energy production and causes various organ disorders. Symptoms due to nuclear DNA are severe, and most cases eventually evolve into Leigh Disease or LBSL. Symptoms due to mtDNA are much less severe. This disease is caused by abnormalities in either nuclear DNA or mtDNA. |
| Complex II Deficiency http://omim.org/entry/252011 |
<Respiratory chain complex abnormalities> Mitochondrial respiratory chain complex II (succinate-ubiquinone oxidoreductase) directly connects the citric acid cycle and the respiratory chain. This disease is very rare and is only caused by nuclear DNA abnormalities. |
| Complex III Deficiency http://omim.org/entry/615158 |
<Respiratory chain complex abnormalities> Low activity of mitochondrial respiratory chain complex III (ubiquinol-cytochrome-c reductase) decreases energy production and causes seizures, hearing loss, and cardiomyopathy. This disease is caused by abnormalities in either nuclear DNA or mtDNA. |
| Complex IV Deficiency / COX Deficiency http://omim.org/entry/220110 |
<Respiratory chain complex abnormalities> Low activity of mitochondrial respiratory chain complex IV (cytochrome c oxidase) decreases energy production. The most common form is Leigh disease. This disease is caused by abnormalities in either nuclear DNA and mtDNA. |
| Complex V Deficiency http://omim.org/entry/604273 |
<Respiratory chain complex abnormalities> Low activity of mitochondrial respiratory chain complex V (ATP synthase) decreases energy production. This disease is caused by abnormalities in nuclear DNA and mtDNA. |
| Fumarase deficiency http://omim.org/entry/606812 |
<Citric acid cycle abnormalities> Impaired fumarate to malate conversion causes defective energy production. Patients develop brain malformation, mental retardation, and seizures. This disease is caused by abnormalities in nuclear DNA. |
| KGDHC Deficiency: α-ketoglutarate Dehydrogenase Complex Deficiency http://omim.org/entry/126063 |
<Citric acid cycle abnormalities> Impaired α-ketoglutarate to succinyl-CoA conversion causes defective energy production, hyperuricemia, and hyperketonemia. Patients develop mental retardation, muscular hypotonia, involuntary movements (erratic and strange movements without conscious thought), seizures, encephalopathy, and metabolic acidosis. This disease is caused by abnormalities in nuclear DNA. particularly in theα-KG gene. |
| Succinyl-CoA Ligase Deficiency http://omim.org/entry/603921 |
<Citric acid cycle abnormalities> Impaired succinyl-CoA to succinate conversion and impaired mtDNA synthesis causes prominent lactic acidosis and lethal infantile mitochondrial disease (LIMD). Patients develop MTDPS, and their imaging findings are similar to those from Leigh disease. This disease is caused by abnormalities in nuclear DNA, particularly in the SUCL gene. |
| PDHC : Pyruvate Dehydrogenase Complex Deficiency http://omim.org/entry/312170 |
<Glucose metabolism abnormalities> Impairment in the process that decomposes carbohydrates and produces energy (pyruvate to acetyl-CoA conversion) causes a lack of energy and metabolic acidosis because unused pyruvate turns into lactic acid. Patients commonly develop psychomotor development disorder, seizures, muscular hypotonia, and respiratory compromise. Complications of Leigh disease are common in boys, while those of microcephaly and hydrocephalus, as well as West syndrome, are common in girls. This disease is caused by abnormalities in nuclear DNA, particularly in the Ela gene. |
| PCD : Pyruvate Carboxylase Deficiency http://omim.org/entry/266150 |
<Glucose metabolism abnormalities> Impaired gluconeogenesis, the pyruvate to oxaloacetate conversion, causes hypoglycemia and inhibits oxaloacetate production. Therefore, patients develop hyperuricemia and produce energy defectively. The degradation pathway for ammonia in the urea cycle is also impaired, and hyperammonemia and hypercitrullinemia occur. Defective production of important brain neurotransmitters, including glutamic acid, causes psychomotor retardation. This disease is caused by abnormalities in nuclear DNA, particularly in the PC gene. |
| CPT I Deficiency: Carnitine palmitoyltransferase I deficiency http://omim.org/entry/255210 |
<Fatty acid metabolism abnormalities> Deficiency in CPT I, which exists in the mitochondrial outer membrane, occurs. CPT I transfers fatty acyl-CoA into the mitochondria and combines acyl-CoA and carnitine. This deficiency affects beta-oxidation of fatty acids, and patients develop seizures and have impaired consciousness due to ketotic hypoglycemia triggered by hunger and infections. This disease is caused by abnormalities in nuclear DNA, particularly in the CPT1 gene. |
| CPT II Deficiency: Carnitine palmitoyltransferase II deficiency http://omim.org/entry/600650 |
<Fatty acid metabolism abnormalities> Abnormalities occur in this enzyme, which exchanges carnitine for acylcarnitine in the mitochondria. This causes impaired oxidation of long-chain fatty acids. The myopathic forms are dominant, whereas the lethal neonatal form and the severe infantile hepatocardiomuscular form are rare. This disease is caused by abnormalities in nuclear DNA, particularly in the CPTII gene. |
| CACT Deficiency: Carnitine-Acyl-Carnitine Deficiency http://omim.org/entry/212138 |
<Fatty acid metabolism abnormalities>The carnitine cycle is an important cycle that imports long-chain fatty acids into the mitochondria. Deficiency in CACT impairs carnitine transportation to and from the mitochondria (reutilization of carnitine) and stops the cycle's function. Therefore, disorders occur in the heart and skeletal muscles, including cardiomyopathy and rhabdomyolysis, which use beta-oxidation as a major energy source. Additionally, impairment of fatty acid utilization in the liver causes symptoms such as nonketotic hypoglycemia and hyperammonemia. This disease is caused by abnormalities in nuclear DNA, particularly in the CACT gene. |
| MCAD: Medium-Chain Acyl-CoA Dehydrogenase Deficiency http://omim.org/entry/201450 |
<Fatty acid metabolism abnormalities> Long-chain fatty acids are beta-oxidized in the mitochondrial inner membrane and then transferred into the middle•short-chain fatty acid beta-oxidation pathway in the matrix. Therefore, MCAD abnormalities affect acetyl-CoA production from acyl-CoA and impair fatty acid beta-oxidation. This disease is caused by abnormalities in nuclear DNA, particularly in the ACADM gene. |
| SCAD: Short-Chain Acyl-CoA Dehydrogenase Deficiency http://omim.org/entry/201470 |
<Fatty acid metabolism abnormalities> SCAD exists in the mitochondrial matrix and catalyzes the first step of beta-oxidation of short-chain acyl-CoA. SCAD deficiency causes mental retardation, delays in language development, muscular hypotonia, impaired growth, seizures, external malformations, hypoglycemic brain damage, microcephaly, optic atrophy, and hypertonicity. This disease is due to the abnormalities in nuclear DNA, particularly in the ACADS gene. |
| SCHAD: Short-Chain-3-hydroxyacyl-CoA Dehydrogenase Deficiency http://omim.org/entry/231530 |
<Fatty acid metabolism abnormalities> HADH exists in the mitochondrial matrix and metabolizes middle•short- chain acyl-CoA. Deficiency of HADH is quite rare and causes encephalopathy, progressive liver disorders, cardiomyopaty, and more. This disease is due to the abnormalities in nuclear DNA (HADH gene abnormalities). |
| Trifunctional Protein Deficiency/Long-chain-hydroxy Acyl-CoA dehydrogenase deficiency http://omim.org/entry/609015 http://omim.org/entry/609016 |
<Fatty acid metabolism abnormalities> Trifunctional protein (TFP) is a group of enzymes that exists in a combined form in the mitochondrial inner membrane. TFP specifically binds to long-chain fatty acids and catalyzes the 2nd, 3rd, and 4th steps of beta-oxidation. Deficiency in these enzymes inhibits long-chain fatty acid beta-oxidation. There is also a case in which only LCHAD (long-chain 3-hydroxyacyl-CoA dehydrogenase) in TFP becomes deficient. This disease is caused by abnormalities in nuclear DNA, particularly in the HADHA and HADHB genes. |
| VLCAD: Very Long-Chain Acyl-CoA Dehydrogenase Deficiency http://omim.org/entry/201475 |
<Fatty acid metabolism abnormalities> VLCAD is an enzyme that exists in combined form in the mitochondrial inner membrane. VLCAD specifically binds to long-chain fatty acids and catalyzes the first step of beta-oxidation. Deficiency in this enzyme impairs long-chain fatty acids beta-oxidation, specifically the acyl-CoA to enoyl-CoA transition. This is caused by abnormalities in nuclear DNA, particularly in the ACADVL gene. |
| Carbamoyl Phosphate Synthetase I deficiency http://omim.org/entry/237300 |
<Amino acid metabolism abnormalities> Disorder in the urea cycle, specifically in the N-acetylglutamic acid to carbamoyl phosphate conversion, inhibits the degradation of ammonia and causes hyperammonemia. Patients develop vomiting, restlessness, and impaired consciousness. This is caused by abnormalities in nuclear DNA, particularly in the CPS1 gene. |
| CCDS3: Cerebral Creatine Deficiency Syndrome 3 http://omim.org/entry/612718 |
<Amino acid metabolism abnormalities> Abnormalities in creatine metabolism, converting arginine and glycine to ornithine and guanidinoacetic acid, respectively, cause creatine deficiency in brain, and patients develop mild psychomotor retardation, seizures, severe language problems, and autism. This is caused by abnormalities in nuclear DNA, particularly in the GATM gene. |
| Citrin Deficiency – NICCD, CTLN2 http://omim.org/entry/603471 http://omim.org/entry/605814 |
<Amino acid metabolism abnormalities> Impaired intramitochondrial transport of aspartate and glutamate causes abnormalities in the urea cycle. Moreover, decreased intake of NADH affects energy production. Patients develop hyperammonemia and citrullinemia. This is caused by abnormalities in nuclear DNA, particularly in the SLC25A13 gene. |
| Hyperprolinemia, type I, type II http://omim.org/entry/239500 http://omim.org/entry/239510 |
<Amino acid metabolism abnormalities> Impaired degradation of proline, involving the conversion of proline to glutamic acid through a P5C intermediate, affects the citric acid cycle. Patients show clinical findings, such as refractory seizures, mental retardation, and acute encephalopathy-like symptoms. This disease is caused by abnormalities in nuclear DNA, particularyly the POX and P5CDH genes, for type I and type II, respectively. |
| GACR: Gyrate Atrophy of Choroid and Retina http://omim.org/entry/258870 |
<Amino acid metabolism abnormalities> The catabolism of ornithine, which is the major intermediate product of the urea cycle, proline, and creatine (ornithine => P5C, glutamic acid), is impaired. Damage occurs, especially in the choroid, resulting in progressive vision loss. This disease is caused by abnormalities in nuclear DNA, particularly in the OAT gene. |
| GCE: Glycine Encephalopathy / Nonketotic Hyperglycinemia http://omim.org/entry/605899 |
<Amino acid metabolism abnormalities> Degradation of glycine, which is a neurotransmitter of the central nervous system, is impaired, and glycine is insufficiently converted to ammonia. Patients develop encephalopathy-like symptoms, such as muscular hypotonia, coma, and seizures within the first few days of life. This is caused by abnormalities in nuclear DNA, particularly genetic deficiencies of GCS. |
| Hyperinsulinemic Hypoglycemia http://omim.org/entry/606762 |
<Amino acid metabolism abnormalities> Excessively secreted insulin, despite low blood glucose, causes hypoglycemia (Glutamic acid is converted to α-ketoglutaric acid, and there is increased citric acid cycle activity). This is caused by abnormalities in nuclear DNA, particularly in the GDH gene. |
| Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome (HHH syndrome) http://omim.org/entry/238970 |
<Amino acid metabolism abnormalities> Impaired transport of ornithine into the mitochondria causes abnormalities in the urea cycle. This syndrome is characterized by intermittent hyperammonemia. This is caused by abnormalities in nuclear DNA, particularly in the ORNT1 gene. |
| N-Acetylglutamate Synthase Deficiency http://omim.org/entry/237310 |
<Amino acid metabolism abnormalities> Impaired urea cycle, specifically the reaction converting glutamic acid and acetyl-CoA into N-acetylglutamic acid, causes hyperammonemia by inhibiting the degradation of ammonia. Patients develop vomiting, restlessness, and impaired consciousness. This is caused by abnormalities in nuclear DNA, particularly in the NAGS gene. |
| MSUD: Maple Syrup Urine Disease http://omim.org/entry/248600 |
<Amino acid metabolism abnormalities> Branched-chain amino acid degradation, convertiing leucine, valine, and isoleucine to succinyl-CoA, is impaired. One type causes serious encephalopathy, while the other type worsens its symptoms, triggered by infections. This is caused by abnormalities in nuclear DNA, particularly in the BCKDH gene. |
| Ornithine Transcarbamylase Deficiency http://omim.org/entry/311250 |
<Amino acid metabolism abnormalities> Impaired urea cycle, specifically the conversion of carbamoyl phosphate to citrulline, inhibits the degradation of ammonia and causes hyperammonemia. Patients develop vomiting, restlessness, and impaired consciousness. This is the most common enzyme deficiency of the urea cycle. It is caused by abnormalities in nuclear DNA, particularly in the OTC gene. |
| Glutaric Acidemia II http://omim.org/entry/231680 |
<Defects in mtDNA maintenance> This disease is mainly caused by impaired fatty acid beta-oxidation due to simultaneous mitochondrial disorders. It is caused by abnormalities in nuclear DNA, particularly in the ETFA and ETFDH genes. |
| Ethylmalonic Encephalopathy http://omim.org/entry/602473 |
<Defects in mtDNA maintenance> Mitochondrial sulfur dioxygenase deficiency increases the level of hydrogen sulfide in the blood and causes minor circulatory impairment. Patients develop CNS, skin, and GI symptoms. This is caused by abnormalities in nuclear DNA, particularly in the ETHE1 gene. |
| L-2-Hydroxyglutaric Aciduria http://omim.org/entry/236792 |
<Defects in mtDNA maintenance> Enzyme deficiency in mitochondrial inner membrane, of which FAD is the coenzyme, inhibits the metabolism of L-2-hydroxyglutaric acid, which is produced from the citric acid cycle and is toxic. Patients develop cerebral leukoencephalopathy. This disease is caused by abnormalities in nuclear DNA, particularly in the L2HGDH gene. |
| (IVA: Isovaleric Acidemia http://omim.org/entry/243500 |
<Defects in mtDNA maintenance> The accumulation of isovaleric acid decreases urea cycle function and energy production. This disease is caused by abnormalities in nuclear DNA, particularly in the IVD gene. |
| 2-Methyl-3-Hydroxybutyryl-Coa Dehydrogenase Deficiency / HSD10 disease http://omim.org/entry/300438 http://omim.org/entry/300256 |
<Defects in mtDNA maintenance> The deficiency of genes required for modification of mitochondrial DNA transcripts causes symptoms similar to those associated with serious mitochondrial encephalopathy. This is caused by abnormalities in nuclear DNA, particularly in the HSD17B10 gene. |
| 3-Methylcrotonyl-Coa Carboxylase 1 Deficiency) http://omim.org/entry/210200 http://omim.org/entry/210210 |
<Defects in mtDNA maintenance>This disease is caused by abnormalities in the catabolism process of leucine, involving the conversion of 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA. Patients develop the disease with symptoms similar to Reye's syndrome, which is triggered by infections. This is caused by abnormalities in nuclear DNA, particularly in the Mcc gene. |
| 3-Methylglutaconic Aciduria http://omim.org/entry/250950 http://omim.org/entry/302060 http://omim.org/entry/258501 http://omim.org/entry/250951 http://omim.org/entry/610198 |
<Defects in mtDNA maintenance> This syndrome is characterized by excessive excretion of 3-methylglutaconic acid in urine. Only type I is due to abnormalities in the catabolism process of leucine, and the other types are considered to be caused by abnormalities of mitochondrial proteins. Type II: This type is also called birth syndrome and is characterized by dilated cardiomyopathy and impaired growth. This is caused by abnormalities in nuclear DNA, particularly in the TAZ gene and in mitochondrial cardiolipin transferase. Type III: This type is also called Costeff optic atrophy syndrome and is characterized by optic atrophy and chorea. This is caused by abnormalities in nuclear DNA, particularly in the OP3 gene. Type IV: This type is characterized by hypertrophic cardiomyopathy, brain atrophy, and developmental delay. There are several reports of nuclear DNA abnormalities as well as mtDNA abnormalities. Type V: This type is characterized by conduction disturbance, cardiomyopathy, and cerebellar ataxia. This is caused by abnormalities in nuclear DNA, particularly in the DNAJC19 gene and mitochondrial inner membrane transferase. |
| Mitochondrial HMG-CoA Synthase Deficiency http://omim.org/entry/605911 http://omim.org/entry/600234 |
<Defects in mtDNA maintenance>The ketone body production from fatty acid beta-oxidation is inhibited. Therefore, acetyl-CoA does not get converted to 3HMG-CoA. Though hunger, via glucagon secretion, stress, and cAMP, triggers beta-oxidation of free fatty acids, it cannot produce ketone bodies and causes nonketotic hypoglycemia. Ketones are produced in the liver as alternative energy source for preventing hypoglycemia. This disease is caused by abnormalities in nuclear DNA, particularly in the HMGCS2 gene. |
| Beta-Ketothiolase Deficiency / Mitochondrial Acetoacetyl-CoA Thiolase Deficiency http://omim.org/entry/203750 http://omim.org/entry/607809 |
<Defects in mtDNA maintenance> Impaired isoleucine metabolism and ketone body utilization in extrahepatic tissue causes ketoacidosis. This is caused by abnormalities in nuclear DNA, particularly in the Acat1 gene. |
| Propionic Acidemia http://omim.org/entry/606054 |
<Defects in mtDNA maintenance> Propionic acid accumulates in mitochondria, impairs the urea cycle, specifically the conversion of propionyl-CoA to D- methylmalonyl-CoA, and causes hyperammonemia. Furthermore, glycine degradation inhibition causes hyperglycinemia. This is caused by abnormalities in nuclear DNA, particularly in the PCC gene. |
| Multiple Respiratory Chain Enzyme Deficiencies http://www.rarediseasesnetwork.org/NAMDC/learnmore/diseases.htm#MRCED |
This is a biochemical finding that indicates mtDNA dysfunction and is caused by depletion of mtDNA, multiple deletions of mtDNA, and defects in translation of mtDNA. All these disorders are due to mutations of nuclear DNA, and most cases of reported multiple RC deficiencies are caused by defects of maintenance or translation of the mtDNA. The former group includes defects of nuclear genes involved in replication of the mtDNA and defects of nucleoside salvage necessary to provide dNTP building blocks for mtDNA synthesis. However, multiple respiratory chain deficiencies may also accompany single mtDNA deletions (KSS, CPEO, Pearson syndrome) and mtDNA tRNA mutations (for example, MELAS, MERRF). |
※The above classification of diseases and conditions caused by mitochondrial functional impairment due to inherited genetic abnormalities was compiled by KOINOBORI from its own viewpoints. A defined classification of mitochondrial diseases has not yet been established in Japan or anywhere else around the world.
- OMIM: Online Mendelian Inheritance in Man
- Biochemistry (Lippincott's Illustrated Reviews Series) 5th edition RA Harvey & DR Ferrier.
- Developmental Disorder List by Yuichi Goto and Takeo Kubota
- Handbook for Mitochondrial Disease National Center Hospital Genetic Counseling Unit
- UR-DBMS: University of the Ryukyus-Database for Malformation Syndromes
- UMDF
- NAMDC