
ミトコンドリア病の種類
ミトコンドリアの主要な機能(呼吸鎖複合体の働きやmtDNAからの必要なタンパク質の翻訳を行う事)に大切なタンパク質のうちの、13種類がmtDNAに基づいて作られています。mtDNAに異常を生じると、ミトコンドリア機能が低下しエネルギーの産生ができなくなり、ミトコンドリア病が発症します。
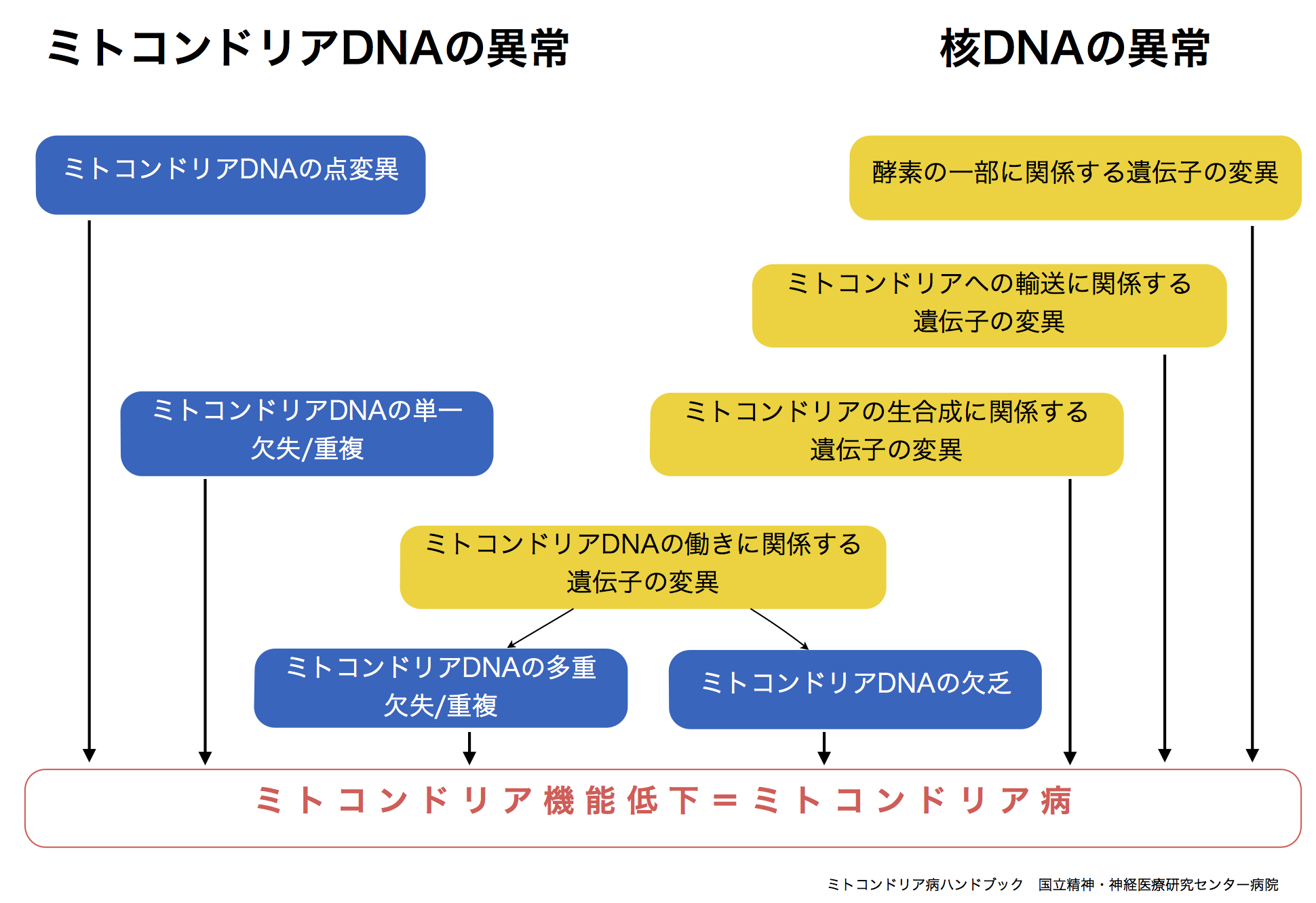
また、ミトコンドリアに必要なタンパク質のうち約1500種類は核DNAの情報に基づき作られています。さらに核DNAはmtDNAの働きそのものにも関与しています。そのため、核DNAに異常があると、ミトコンドリア内のエネルギー代謝(物質の分解合成過程から呼吸鎖複合体におけるエネルギー産生)経路において広範囲にわたり様々な影響が生じ、色々なミトコンドリア病が発症します。
主なミトコンドリア病
ミトコンドリアDNAの異常が主体
| 疾患名 | 原因 | 症状 |
|---|---|---|
| 慢性進行性外眼筋麻痺(CPEO:Chronic Progressive External Ophthalmoplegia) http://omim.org/entry/157640 |
mtDNAの異常 | 目の周りの筋肉が麻痺して眼球を動かせなくなる。これに網膜の変性と心臓の伝導障害を合併する場合、カーンズ・セイヤー症候群(OMIM #530000)と呼ぶ。 |
| ピアソン病(Pearson Syndrome) http://omim.org/entry/557000 |
mtDNAの異常 | 乳児期に鉄芽球性貧血と膵外分泌不全を発症する。幼少時の生存者はカーンズ・セイヤー症候群に移行する。 |
| カーンズ・セイヤー症候群(KSS) http://omim.org/entry/530000 |
mtDNAの異常 | 眼瞼下垂及び外眼筋麻痺、心伝導障害、網膜色素変性を3徴とする症候群である。通常小児期に発症し、病因が極めて類似していることからKSSはCPEOの重症型と考えられる。CPEOの70%、KSSの90%に、mtDNAの異常を認める。 |
| 糖尿病難聴症候群(DAD:Diabetes and Deafness) http://omim.org/entry/520000 |
mtDNAの異常 | 糖尿病の発症と聴力障害を伴う。 |
| レーバー(レーベル)遺伝性視神経萎縮症(LHON:Leber Hereditary Optic Neuropathy) http://omim.org/entry/535000 |
mtDNAの異常 | 主に10歳から30歳くらいに両眼性の視力低下で発症する。その後除除に視神経乳頭の蒼白化が始まり、通常1年程度で視神経萎縮に至る。 |
| レーバー(レーベル)・プラス(LHON-Plus) http://omim.org/entry/500001 |
mtDNAの異常 | LHONの視神経症状に加え、異常な動きや協調運動障害、ジストニア、運動失調、痙縮などの中枢神経症状及び心臓の障害を伴う。 |
| ニューロパシー,運動失調,網膜色素変性症候群(NARP:Neuropathy, Ataxia, and Retinitis Pigmentosa Syndrome) http://omim.org/entry/551500 |
mtDNAの異常 | 神経障害、運動失調、および網膜色素変性症を発症する。MILSとの関連が高い。 |
| 母性遺伝性リー脳症(MILS:Maternally Inherited Leigh Syndrome) http://omim.org/entry/256000 |
mtDNAの異常 | mtDNAの異常によって生じるリー症候群である。 |
| メラス:ミトコンドリア脳筋症・乳酸アシドーシス・脳卒中様発作症候群(MELAS:Mitochondrial Encephalomyopathy Lactic Acidosis and Strokelike Episodes) http://omim.org/entry/540000 |
mtDNAの異常 | 脳卒中様症状を伴うミトコンドリア病で、急激な意識障害や運動麻痺など脳卒中に似た症状が現れるという特徴がある。心臓や膵臓、耳、内分泌器官などの臓器に症状がおこる場合もある。症状の種類や程度は患者ごとに異なり、一人の患者でも時間とともに変化することがある。 |
| マーフ:赤色ぼろ線維を伴うミオクローヌスてんかん(MERRF:Myoclonic Epilepsy and Ragged-Red Fiber Disease) http://omim.org/entry/545000 |
mtDNAの異常 | ミオクローヌスを伴うミトコンドリア病で、主に脳と筋肉に症状が現れる。体がふらついたり(小脳症状)、 自分の意思とは関係なく筋肉が動いたりする (ミオクローヌス) 。 |
| 家族性両側線条体壊死(FBSN:Familial Bilateral Striatal Necrosis)/ ミトコンドリア線条核黒質変性 (Striatonigral Degeneration ) http://omim.org/entry/500003 |
mtDNAの異常 | mtDNA異常によるミトコンドリア呼吸鎖Ⅴ(ATP合成酵素)欠損症である。 |
| ルフト病(Luft Disease) http://omim.org/entry/251900 |
mtDNAの異常 | ミトコンドリア筋症の一つである。発熱、多量の発汗、多食、多飲、赤色ぼろ線維、および頻脈と代謝亢進を伴う。穏やかな弱さと運動不耐性がある。 |
| アミノグリコシド感受性難聴(AID:Aminoglycoside-induced Deafness) http://omim.org/entry/580000 |
mtDNAの異常 | アミノグリコシド系薬剤により引き起こされる全聾である。 |
| ミトコンドリアDNA多量欠失症候群(Multiple Deletions of Mitochondrial DNA) http://www.ncbi.nlm.nih.gov/books/NBK1203 |
mtDNAの異常 | 多様なmtDNAの遺伝的欠陥(少なくとも5核遺伝子が知られている)が原因で、通常はCPEOが類似の臨床症状を持つ。関連する症状は、難聴、筋障害、運動失調、末梢神経障害、構音障害、視神経萎縮、認知症、パーキンソニズムなどがある。 |
核DNAの異常が主体
| 疾患名 | 原因 | 症状 |
|---|---|---|
| アルパース症候群、乳児進行性脳灰白質ジストロフィー(Alpers syndrome) http://omim.org/entry/203700 |
ミトコンドリアDNA保持不全 | 痙攣発作、認知症、痙縮、失明、肝機能障害、および脳変性症などを認める。核DNA突然変異(POLG遺伝子:mtDNA複製酵素)に起因し、POLG遺伝子変異の最重症表現型である。 |
| ミトコンドリア劣性運動失調症候群(MIRAS:Mitochondrial Recessive Ataxia Syndrome) http://omim.org/entry/607459 |
mtDNA保全の異常 | SANDOとともに現在は、運動失調ニューロパチースペクロラム(ANS)としてPOLG関連疾患となっている。 |
| ミトコンドリア神経胃腸管性脳症症候群(MNGIE:Mitochondrial Neurogastrointestinal Encephalomyopathy) http://omim.org/entry/603041 |
mtDNA保全の異常 | チミジンホスホリラーゼが欠損するとDNAを作る際に重要な核酸代謝に異常が生じる。分裂している細胞は少ないが、mtDNAの複製は頻度が高いので、mtDNAの量や質の多大な障害が起きる。MTDPSの1つである。CPEO、末梢神経障害、消化管障害、白質脳症、悪液質(極端な消耗)、乳酸アシドーシス、赤色ぼろ繊維などを認める。 核DNA異常(チミジンホスホリラーゼをコードするTYMP遺伝子の変異)に起因する。 |
| ミトコンドリアDNA枯渇症候群(MTDPS:Mitochondrial DNA Depletion Syndrome) http://omim.org/entry/603041 http://omim.org/entry/251880 |
mtDNA保全の異常 | mtDNAの構成分子に関わる遺伝子に異常を認める多様な遺伝子疾患である。ミオパチー(乳児期や子供の頃に発症する筋原性疾患)や肝脳疾患の主要な原因となる。 |
| POLG関連疾患(POLG mutation) http://omim.org/entry/174763 http://omim.org/entry/613662 http://omim.org/entry/604983 http://omim.org/entry/203700 http://omim.org/entry/157640 http://omim.org/entry/258450 http://omim.org/entry/607459 http://omim.org/entry/606075 http://omim.org/entry/609283 http://omim.org/entry/609286 http://omim.org/entry/610131 http://omim.org/entry/603041 http://omim.org/entry/604419 |
mtDNA保全の異常 | ・アルパース 症候群 (AHS):最重症表現型の1つで重度の脳症となる。 ・小児期筋大脳肝症スペクトラム (MCHS):発達遅滞または認知症, 乳酸性アシドーシス, 発育不全を伴うミオパチーがみられる。 ・ミオクローヌスてんかん-ミオパチー-感覚性運動失調 (MEMSA) :てんかん, ミオパチー, 眼球運動麻痺のない運動失調のスペクトラムを有する。以前の脊髄小脳失調-てんかん (SCAE) を含む。 ・運動失調ニューロパチースペクトラム (ANS):以前のミトコンドリア性劣性運動失調症候群 (MIRAS) と感覚性運動失調ニューロパチー-構音障害と眼球運動麻痺 (SANDO) を含む。 ・常染色体劣性進行性外眼筋麻痺 (arPEO) :進行性外眼筋衰弱が特徴で, 眼瞼下垂と眼筋不全麻痺となる (全身病変はない)。 ・常染色体優性進行性外眼筋麻痺 (adPEO):典型的には全身性ミオパチーを含み, いろんな程度の感音難聴, 軸索ニューロパチー, 運動, うつ, パーキンソン病, 性腺機能低下症, および白内障がみられる 。 |
| 感覚性運動失調性ニューロパチー -構音障害-眼球運動不全麻痺(SANDO:Sensory Ataxia Neuropathy Dysarthria Opthalmoplegia) http://omim.org/entry/174763 |
mtDNA保全の異常 | MIRASとともに現在は運動失調ニューロパチースペクロラム(ANS)としてPOLG関連疾患となっている。 |
| 白質脳症(LBSL: Leukoencephalopathy with Brainstem and Spinal cord involvement and Lactate elevation) http://omim.org/entry/611105 |
mtDNAの翻訳の障害 | ミトコンドリアアスパルチルtRNA 合成酵素欠損症による脳幹及び脊髄病変である。乳酸高値、筋萎縮、筋衰弱、運動失調、振戦などが生じる。核DNAの異常(DARS2遺伝子異常)に起因する |
| コエンザイムQ10欠乏症(Co-Enzyme Q10 Deficiency) http://omim.org/entry/607426 |
脂質膜の異常 | 脳筋症、精神遅滞、運動不耐性、赤色ぼろ線維、および尿中の再発性ミオグロビンが生じる。核DNAの異常に起因する。 |
| リー症候群、リー脳症、リー病(Leigh Disease or Syndrome) http://omim.org/entry/256000 http://omim.org/entry/308930 |
呼吸鎖複合体主体の異常 | 2歳以前から始まる精神運動発達遅延、血中や髄液中の乳酸・ピルビン酸高値、CTあるいはMRIにおける大脳基底核や脳幹の対称性の壊死性病変を特徴とする。初発症状は、食事摂取障害がもっとも多く、ついで運動発達遅延や体腔、筋緊張低下である。眼球運動の異常、視神経萎縮、視力障害などの眼の症状、けいれん、呼吸障害、小脳症状などをみる。少なくとも26個の遺伝子異常が同定されており、それらの異常はピルビン酸デヒドロゲナーゼ(PDHC)欠損症、および呼吸鎖酵素(複合体I、II、IV、およびV)の欠損症に関係する。核DNAの異常に起因する。 |
| 呼吸鎖複合体 I 欠損症(Complex I Deficiency) http://omim.org/entry/252010 |
呼吸鎖複合体の異常 | ミトコンドリア呼吸鎖複合体Ⅰ(NADH−ユビキノン酸化還元酵素)の活性低下によりエネルギー産生が低下し、各臓器に障害が起きる。核DNA由来の場合は症状が酷く最終的にはほとんどがリー症候群や白質脳症に移行する。mtDNA由来の場合ははるかに軽症である。核DNA異常とmtDNA異常がある。 |
| 呼吸鎖複合体Ⅱ欠損症(Complex ⅡDeficiency) http://omim.org/entry/252011 |
呼吸鎖複合体の異常 | ミトコンドリア呼吸鎖複合体Ⅱ(コハク酸−ユビキノン酸化還元酵素)はクエン酸回路と呼吸鎖を直接つないでいる。極めてまれであり、核DNA異常のみがある。 |
| 呼吸鎖複合体Ⅲ欠損症(Complex ⅢDeficiency) http://omim.org/entry/615158 |
呼吸鎖複合体の異常 | ミトコンドリア呼吸鎖複合体Ⅲ(ユビキノール−シトクロムC還元酵素)の活性低下によりエネルギー産生が低下する。けいれん、難聴、心筋症などが生じる。核DNA異常とmtDNA異常がある。 |
| 呼吸鎖複合体 Ⅳ欠損症 / COX欠損症(Complex Ⅳ Deficiency / COX Deficiency) http://omim.org/entry/220110 |
呼吸鎖複合体の異常 | ミトコンドリア呼吸鎖複合体Ⅳ(シトクロムC酸化酵素)の活性低下によりエネルギー産生が低下する。最も頻度の高い病型はリー症候群である。核DNA異常とmtDNA異常がある。 |
| 呼吸鎖複合体 V 欠損症(Complex V Deficiency) http://omim.org/entry/604273 |
呼吸鎖複合体の異常 | ミトコンドリア呼吸鎖複合体Ⅴ(ATP合成酵素)の活性低下によりエネルギー産生が低下する。核DNA異常とmtDNA異常がある。 |
| フマラーゼ欠損症(Fumarase deficiency) http://omim.org/entry/606812 |
クエン酸回路の異常 | フマル酸⇒リンゴ酸に障害が生じる。エネルギーの産生障害が生じ、脳の奇形や精神運動の発達の遅れ、けいれんなどが起きる。核DNAの異常がある。 |
| α-ケトグルタル酸脱水素酵素複合体欠損症(KGDHC Deficiency:α-ketoglutarate Dehydrogenase Complex Deficiency) http://omim.org/entry/126063 |
クエン酸回路の異常 | αケトグルタル酸⇒スクシニルCoAに障害が生じる。エネルギーの産生障害と高尿酸血症、高ケトン血症が生じ、精神運動発達の遅れ、筋緊張低下、不随意運動(意識しないのに起きる不規則で奇妙な動き)やけいれん、脳症、代謝性アシドーシスなどが生じる。核DNAの異常(α-KG遺伝子異常)に起因する。 |
| スクシニルCoAリガーゼ欠損症(Succinyl-CoA Ligase Deficiency) http://omim.org/entry/603921 |
クエン酸回路の異常 | スクシニルCoA⇒コハク酸とmtDNAの合成に障害が生じる。著明な乳酸アシドーシスをきたし乳児致死型ミトコンドリア病(LIMD)を生じる。MTDPSをきたし、画像所見上もリー脳症類似の所見が見られる。核DNAの異常(SUCL遺伝子異常)に起因する。 |
| ピルビン酸脱水素酵素複合体欠損症(PDHC : Pyruvate Dehydrogenase Complex Deficiency) http://omim.org/entry/312170 |
糖代謝の異常 | 糖質を分解してエネルギー産生する過程(ピルビン酸⇒アセチルCoA)に障害が生じる。エネルギー不足と代謝性アシドーシス(使われないピルビン酸が乳酸になる)がおきる。精神運動発達障害、けいれん、筋緊張低下、呼吸障害が一般的にみられ、男児ではリー症候群の合併、女児では小頭症、水頭症、West症候群の合併が多い。核DNAの異常(Ela遺伝子異常)がある。 |
| ピルビン酸カルボキシラーゼ欠損症(PCD : Pyruvate Carboxylase Deficiency) http://omim.org/entry/266150 |
糖代謝の異常 | 糖新生(ピルビン酸⇒オキサロ酢酸)に障害を生じ低血糖、オキサロ酢酸ができないために、エネルギー産生障害、高乳酸血症がおきる。さらにアンモニアの分解する経路(尿素回路)も障害を受けるため、高アンモニア血症や高シトルリン血症が起きる。また脳内の重要な神経伝達物質(グルタミン酸)も作れなくなるので精神運動発達が遅れる。核DNAの異常(PC遺伝子異常)がある。 |
| カルニチン・パルミトイルトランスフェラーゼ I 欠損症(CPT I Deficiency:Carnitine palmitoyltransferase I deficiency) http://omim.org/entry/255120 |
脂肪酸代謝の異常 | ミトコンドリア外膜から脂肪酸アシルCoAをミトコンドリア内に輸送する為に、ミトコンドリア外膜に存在しアシルCoAとカルニチンを結合させる酵素の欠損症である。脂肪酸β酸化に障害がでるため、空腹時や感染症を契機にけいれんや意識障害(低ケトン性低血糖)が生じる。核DNAの異常(CPT1遺伝子異常)がある。 |
| カルニチン・パルミトイルトランスフェラーゼ Ⅱ欠損症(CPTⅡ Deficiency:Carnitine palmitoyltransferase Ⅱ deficiency) http://omim.org/entry/600650 |
脂肪酸代謝の異常 | ミトコンドリア内に取り込まれたアシルカルニチンからカルニチンを切り離す酵素に異常がでるので、長鎖脂肪酸酸化障害を引き起こす。多くを占める筋型とまれにある致死性新生児型と重症乳児肝心筋症がある。核DNAの異常(CPTⅡ遺伝子異常)がある。 |
| カルニチン・アシル・カルニチントランスロカーゼ欠損症(CACT Deficiency : Carnitine-Acyl-Carnitine Deficiency) http://omim.org/entry/212138 |
脂肪酸代謝の異常 | カルニチン回路は長鎖脂肪酸をミトコンドリアに取込む重要な回路であり、本酵素が欠損すると、カルニチンの膜内外への輸送ができず(カルニチンの再利用ができず)、この回路が働かなくなる。従ってβ酸化を主なエネルギーとしている心臓、骨格筋の異常(心筋症、横紋筋融解)が生じ、また肝臓での脂肪酸利用障害から非ケトン性低血糖、高アンモニア血症などが生じる。核DNAの異常(CACT遺伝子異常)に起因する。 |
| 中鎖アシル-CoA脱水素酵素欠損症(MCAD:Medium-Chain Acyl-CoA Dehydrogenase Deficiency) http://omim.org/entry/201450 |
脂肪酸代謝の異常 | ミトコンドリア内膜での長鎖脂肪酸のβ酸化の後にマトリックスでの中鎖・短鎖β酸化回路に移る。従ってMCADの異常によりアシルCoAからアセチルCoAの産生が障害され、脂肪酸β酸化が出来なくなる。核DNA異常(ACADM遺伝子)に起因する。 |
| 短鎖アシル-CoA脱水素酵素欠損症(SCAD:Short-Chain Acyl-CoA Dehydrogenase Deficiency) http://omim.org/entry/201470 |
脂肪酸代謝の異常 | SCADはミトコンドリアマトリックスに存在して短鎖アシルCoAのβ酸化の最初のステップを触媒している。精神発達遅滞、言葉の遅れ、筋緊張低下、成長障害、けいれん、外形奇形、低血糖性脳障害、小頭症、視神経萎縮、筋緊張亢進などが生じる。核DNAの異常( ACADS遺伝子異常)がある。 |
| 3-ヒドロキシアシル-CoA脱水素酵素(HADH)欠損症 / SCHAD:Short-Chain-3-hydroxyacyl-CoA Dehydrogenase Deficiency http://omim.org/entry/231530 |
脂肪酸代謝の異常 | HADHはミトコンドリアマトリックスに存在し、中鎖・短鎖アシルCoAの代謝を担当している。脳症や進行性肝障害、心筋症等を生じ極めて稀である。核DNAの異常( HADH遺伝子異常)に起因する。 |
| ミトコンドリア三頭酵素欠損症/長鎖-3-ヒドロキシアシルCoA脱水素酵素欠損症(Trifunctional Protein Deficiency) http://omim.org/entry/609015 http://omim.org/entry/609016 |
脂肪酸代謝の異常 | ミトコンドリア三頭酵素(TFP)はミトコンドリア内膜に結合した形で存在し、長鎖脂肪酸特異的に結合しβ酸化の第2・3・4ステップを触媒する酵素群である。本酵素の欠損にて長脂肪酸のβ酸化が阻害される。TFPの中で、LCHAD:Long-chain 3-hydroxyacyl-CoA dehydrogenase(長鎖-3-ヒドロキシアシルCoA脱水素酵素)のみが欠損する場合(LCAD欠損症)もある。核DNAの異常(HADHA, HADHB遺伝子異常)に起因する。 |
| 極長鎖アシル-CoA脱水素酵素欠損症(VLCAD:Very Long-Chain Acyl-CoA Dehydrogenase Deficiency) http://omim.org/entry/201475 |
脂肪酸代謝の異常 | VLCADはミトコンドリア内膜に結合した形で存在し、長鎖脂肪酸特異的に結合しβ酸化の第一ステップを触媒する酵素である。本酵素の欠損にて長鎖脂肪酸のβ酸(アシルCoA⇒エノイルCoA)が障害される。核DNAの異常(ACADVL遺伝子異常)。 |
| カルバモイルリン酸合成酵素Ⅰ欠損症(Carbamoyl Phosphate Synthetase Ⅰ deficiency) http://omim.org/entry/237300 |
アミノ酸代謝の異常 | 尿素回路(N-アセチルグルタミン酸⇒カルバモイルリン酸)に障害が生じる。アンモニアの分解ができないので高アンモニア血症をきたし、嘔吐、不穏、意識障害を起こす。核DNAの異常(CPS-1遺伝子異常)。 |
| クレアチン代謝異常(CCDS3:Cerebral Creatine Deficiency Syndrome 3) http://omim.org/entry/612718 |
アミノ酸代謝の異常 | クレアチン代謝(アルギニン、グリシン⇒オルニチン、グアニジドアセト酢酸)異常では脳内のクレアチンが欠乏し軽度の精神運動の遅れ、けいれん、重度の言語障害や自閉症状が見られる。核DNAの異常(GATM遺伝子異常)がある。 |
| シトリン欠損症–NICCDとCTLN2(Citrin Deficiency) http://omim.org/entry/603471 http://omim.org/entry/605814 |
アミノ酸代謝の異常 | ミトコンドリア内のアスパラギン酸グルタミン酸輸送に障害が生じ、尿素回路に異常が起きる。またNADHの取込みが減少するため、エネルギー産生に障害も起きる。高アンモニア血症、シトリン血症を認める。核DNAの異常(SLC25A13遺伝子異常)がある。 |
| 高プロリン血症Ⅰ型・Ⅱ型(Hyperprolinemia, type I, typeⅡ) http://omim.org/entry/239500 http://omim.org/entry/239510 |
アミノ酸代謝の異常 | プロリンの分解(プロリン⇒P5C⇒グルタミン酸)が障害されクエン酸回路に支障がでる。難治性のけいれんや精神発達の遅れ、急性脳症様の所見を示す。核DNAの異常(Ⅰ型:POX、Ⅱ型:P5CDH遺伝子異常)に起因する。 |
| 脳回転状脈絡膜網膜萎縮症(GACR:Gyrate Atrophy of Choroid and Retina) http://omim.org/entry/258870 |
アミノ酸代謝の異常 | 尿素回路、プロリン、クレアチンの主要な中間産物であるオルニチンの異化反応(オルニチン⇒P5C、グルタミン酸)に障害が生じる。特に脈絡膜に障害が強く現れ進行性の視力障害をきたす。核DNAの異常(OAT遺伝子異常)がある。 |
| 非ケトーシス型高グリシン血症(GCE:Glycine Encephalopathy) http://omim.org/entry/605899 |
アミノ酸代謝の異常 | 中枢神経の神経伝達物質であるグリシンの分解(グリシン⇒アンモニア)が障害され、生後数日以内に発生する筋緊張低下、昏睡、けいれんなどの脳症様症状をきたす。核DNAの異常(GCSの遺伝的欠損)がある |
| グルタミン酸脱水素酵素異常症(Hyperinsulinemic Hypoglycemia) http://omim.org/entry/606762 |
アミノ酸代謝の異常 | 血糖が低いにも関わらずインシュリンが過剰に分泌し低血糖が生じる(グルタミン酸⇒α–ケトグルタル酸↑⇨クエン酸回路↑)。核DNAの異常(GDH遺伝子異常)がある。 |
| 高オルニチン血症・高アンモニア血症・ホモシトルリン尿症症候群(HHH syndrome) http://omim.org/entry/238970 |
アミノ酸代謝の異常 | オルニチンのミトコンドリア膜輸送が障害され、尿素回路に異常が生じる。間欠的な高アンモニア血症が特徴である。核DNAの異常(ORNT1遺伝子異常)がある。 |
| N–アセチルグルタミン酸合成酵素欠損症(N-Acetylglutamate Synthase Deficiency) http://omim.org/entry/237310 |
アミノ酸代謝の異常 | 尿素回路(グルタミン酸+アセチルCoA⇒N-アセチルグルタミン酸))に障害が生じる。アンモニアの分解ができないので高アンモニア血症をきたし、嘔吐、不穏、意識障害を起こす。核DNAの異常(NAGS遺伝子異常)がある。 |
| メープルシロップ尿症(MSUD:Maple Syrup Urine Disease) http://omim.org/entry/248600 |
アミノ酸代謝の異常 | 分岐鎖アミノ酸の分解過程(ロイシン、バリン、イソロイシン⇒スクシニルCoA)に障害が起きる。重篤な脳症を示すタイプと感染症を契機に悪化するタイプがある。核DNAの異常(BCKDH遺伝子異常)がある。 |
| オルニチントランスカルバミラーゼ欠損症(OrnithineTranscarbamylase Deficiency) http://omim.org/entry/311250 |
アミノ酸代謝の異常 | 尿素回路(カルバモイルリン酸⇒シトルリン)に障害が生じる。アンモニアの分解ができないので高アンモニア血症をきたし、嘔吐、不穏、意識障害を起こす。尿素回路の酵素欠損では最多である。核DNAの異常(OTC遺伝子異常)がある。 |
| グルタル酸血症Ⅱ型(Glutaric Acidemia Ⅱ) http://omim.org/entry/231680 |
有機酸代謝障害 | ミトコンドリアの複数の脱水素酵素が同時に障害され、脂肪酸β酸化障害を主体とする。核DNAの異常(ETFA, ETFDH遺伝子異常)に起因する。 |
| エチルマロン酸脳症(Encephalopathy) http://omim.org/entry/602473 |
有機酸代謝障害 | ミトコンドリアの硫黄ジオキシゲナーゼの欠損により、血液中の硫化水素の濃度が増加する。微小循環障害が生じ、中枢、皮膚、消化管症状が起きる。核DNAの異常(ETHE1遺伝子)がある。 |
| L-2-ヒドロキシグルタル酸尿症(L-2-Hydroxyglutaric Aciduria) http://omim.org/entry/236792 |
有機酸代謝障害 | ミトコンドリア内膜に存在する酵素(FADを補酵素)で欠損することにより、クエン酸回路とリンクして産生される毒性のあるL-2-ヒドロキシグルタル酸を処理できなり、大脳白質障害を生じる。核DNAの異常(L2HGDH遺伝子異常)に起因する。 |
| イソ吉草酸血症(IVA:Isovaleric Acidemia) http://omim.org/entry/243500 |
有機酸代謝障害 | イソ吉草酸の蓄積により尿素回路機能低下やエネルギー産生の低下を来す。核DNAの異常(IVD遺伝子異常)に起因する。 |
| 2-メチル-3-ヒドロキシブチリルCoA脱水素酵素欠損症(HSD10病)(2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency) http://omim.org/entry/300438 http://omim.org/entry/300256 |
有機酸代謝障害 | ミトコンドリアDNAからの転写物の修飾に必要な遺伝子の欠損のため、重篤なミトコンドリア脳症に類似する。核DNA異常(HSD17B10遺伝子)がある。 |
| 3-メチルクロトニルCoAカルボキシラーゼ欠損症(3-Methylcrotonyl-Coa Carboxylase 1 Deficiency) http://omim.org/entry/210200 http://omim.org/entry/210210 |
有機酸代謝障害 | ロイシンの異化過程(3-メチルクロトニルCoA⇒3-メチルグルタコニルCoA)の異常。感染症などを契機にライ症候群に似た症状で発症する。核DNAの異常(Mcc遺伝子異常)がある。 |
| メチルグルタコン酸尿症(3-Methylglutaconic Aciduria) http://omim.org/entry/250950 http://omim.org/entry/302060 http://omim.org/entry/258501 http://omim.org/entry/250951 http://omim.org/entry/610198 |
有機酸代謝障害 | 尿中の3-メチルグルタコン酸の過剰排泄を特徴とする症候群。ロイシン異化過程の異常で生じるのはⅠ型のみで、他の型はミトコンドリアに関連する蛋白の異常が原因と考えられる。 Ⅱ型:Birth症候群とも言われる。拡張型心筋症と成長障害を伴う。核DNAの異常(TAZ遺伝子:ミトコンドリアカルジオリピン転移酵素)がある。 Ⅲ型:Costeff視神経萎縮症とも言われる。視神経萎縮、舞踏様運動を伴う。核DNAの異常(OP3遺伝子)がある。 Ⅳ型:肥大型心筋症、脳萎縮、発達の遅れを伴う。核DNA異常、mtDNAの異常など報告が複数ある。 Ⅴ型:伝導障害、心筋症、小脳失調を伴う。核DNAの異常(DNAJC19遺伝子:ミトコンドリア内膜転移酵素)がある。 |
| ミトコンドリアHMG-CoA合成酵素欠損症(Mitochondrial HMG-CoA Synthase Deficiency) http://omim.org/entry/605911 http://omim.org/entry/600234 |
有機酸代謝障害 | 脂肪酸β酸化からのケトン体産生(アセチルCoA⇒3HMG-CoA)ができなくなる。空腹(グルカゴン分泌、ストレス(cAMP)にて遊離脂肪酸が動員されβ酸化は行われるがケトン体が産生できない為、非ケトン性低血糖となる。ケトン体は肝臓にて低血糖を防ぐ為の代替エネルギーとして産生される。核DNAの異常(HMGCS2遺伝子)がある。 |
| β−ケトチオラーゼ(ミトコンドリア・アセトアセチルCoAチオラーゼ欠損症)(Mitochondrial Acetoacetyl-CoA Thiolase Deficiency) http://omim.org/entry/203750 http://omim.org/entry/607809 |
有機酸代謝障害 | イソロイシン代謝とケトン体の肝外組織における利用が障害され、ケトアシドーシス発作を起こす。核DNAの異常(Acat1遺伝子異常)がある。 |
| プロピオン酸血症(Propionic Acidemia) http://omim.org/entry/606054 |
有機酸代謝障害 | ミトコンドリア内に蓄積したプロピオン酸により尿素回路(プロピオニルCoA⇨D-メチルマロニルCoA)が障害され高アンモニア血症が生じ、またグリシン分解が阻害され高グリシン血症を生じる。核DNAの異常(PCC遺伝子異常)がある。 |
| 多ミトコンドリア呼吸鎖酵素欠損(Multiple Respiratory Chain Enzyme Deficiencies) http://www.rarediseasesnetwork.org/NAMDC/learnmore/diseases.htm#MRCED |
その他 | mtDNAの枯渇、複数のmtDNAの欠失またはmtDNAの翻訳の欠陥が原因で生じる、生化学的所見としてのmtDNAの機能不全を示すものである。全てのこれらの障害は、核DNAにおける突然変異によるものであり、報告された複数のRC欠陥のほとんどの症例は、mtDNAの維持または翻訳の欠陥に起因しているmtDNAの複製とmtDNA合成に必要な核酸の構成物質に関わる核DNAの欠陥が含まれている。しかし、複数の呼吸鎖の欠陥は、単一のmtDNAの欠失(KSS、CPEO、ピアソン症候群)とのmtDNAのtRNA変異(例えば、MELAS、MERRF)を伴うことがある。 |
※上記分類は、先天的な遺伝子異常によるミトコンドリアの機能障害により引き起こされる疾患及び病態について、“こいのぼり”が独自でまとめたものです。日本のみならず世界において、ミトコンドリア病の分類について定められたものは、未だ存在しません。
- OMIM : Online Mendelian Inheritance in Man (http://omim.org)
- イラストレイテッド 生化学 5th edition RA Harvey & DR Ferrier.
- 発達障害疾患リスト 後藤雄一, 久保田健夫(http://www.ncnp.go.jp/nin/guide/r2/genedigmanu_html/listABC.html)
- ミトコンドリア病ハンドブック 国立精神・神経医療研究センター病院 遺伝カウンセリング室
- UR-DBMS:琉球大学遺伝学性疾患データベース
- UMDF (http://www.umdf.org/site/pp.aspx?c=8qKOJ0MvF7LUG&b=7934629)
- NAMDC (https://rarediseasesnetwork.epi.usf.edu/NAMDC/learnmore/diseases.htm)
- 先天代謝異常ハンドブック 遠藤文夫
- Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviewsTM 2010